精確的非共價相互作用能量數據對於構建生物大分子系統的分子動力學模擬力場至關重要。在構建一個可靠的力場時,有兩個重要的實際問題需要考慮,希望能平衡所需的化學精度和工作效率。一個是確定適合計算相互作用能量的量子化學理論級別;另一個是使用合適的連續能量函數來建模量子化學能量數據。對於第一個問題,我們最近使用SAPT0理論級別計算了分子間相互作用能量,並系統地將這些能量組織成ab initio SOFG-31(同源二聚體)和SOFG-31-異源二聚體數據集。在這項工作中,我們使用更先進的SAPT2理論級別和更廣泛的基組系列重新計算這些相互作用能量(表- 1、表- 2、表- 3)。我們的目的是確定相對於CCSD(T)/CBS基準化學精度的相互作用能量適合的SAPT理論級別。接下來,為了利用這些能量數據集,我們採用了一種成熟的機器學習技術,稱為CLIFF方案,來構建一個通用的生物分子動力學模擬力場。這裡我們使用SOFG-31數據集作為訓練集,使用SOFG-31-異質二聚體數據集作為測試集。我們的結果顯示,使用CLIFF方案只需一個小的訓練集就能重現多種二聚體相互作用能量模式。每個SAPT能量組分以及SAPT總能量的整體誤差都遠低於所需的化學精度~1 kcal/mol(表- 4)。(應力所趙聖德教授提供)
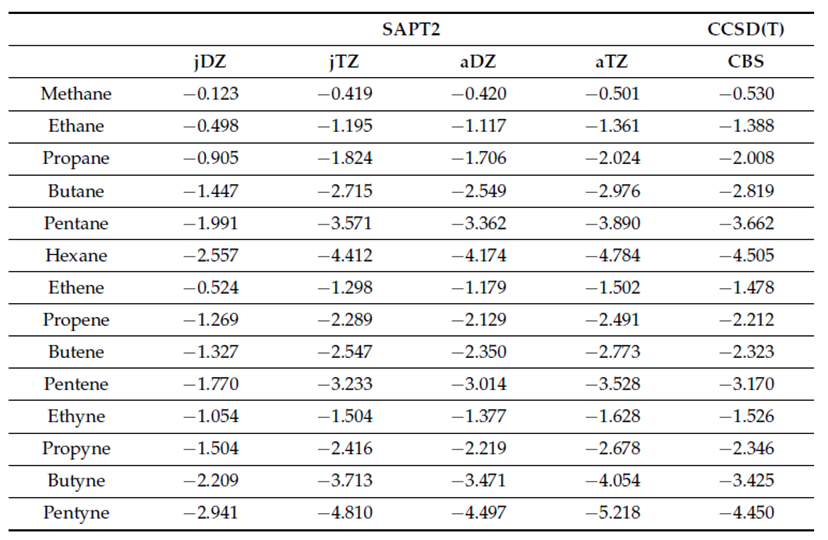
表- 1 SAPT2計算的AaAeAy組二聚體的相互作用能量(以kcal/mol為單位)。
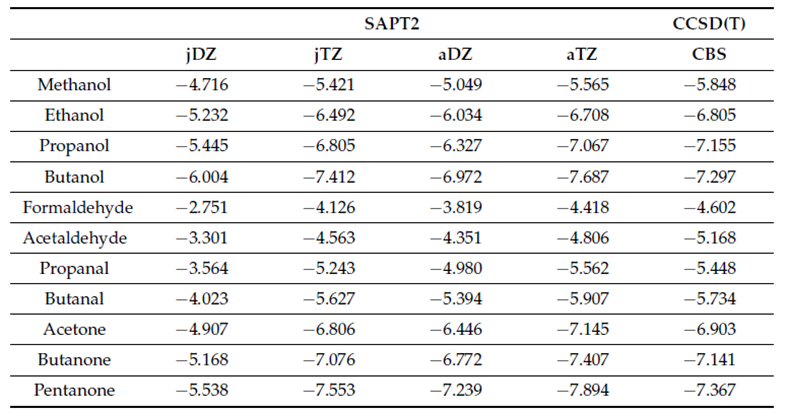
表- 2 SAPT2 計算的 AcAdK 組二聚體的相互作用能量(以kcal/mol為單位)。
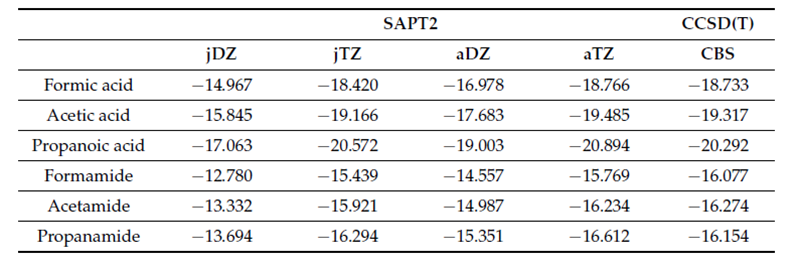
表- 3 SAPT2 計算的 CAA 組二聚體的相互作用能量。
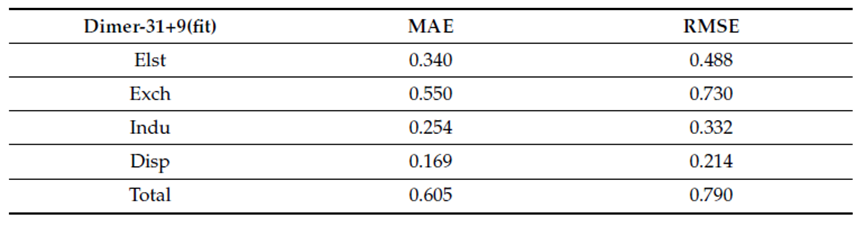
表- 4使用 Dimer-31+9 集合來預測 SOFG-31-異源二聚體的結果。